
Project C2
Doctoral researcher: Gayatri Jagirdar
Principle investigator: Uwe Lendeckel
Co-supervisors: U. Bornscheuer, F. Scholz, O. Otto
Impact of Tafazzin knock-out on cellular and mitochondrial function
Background
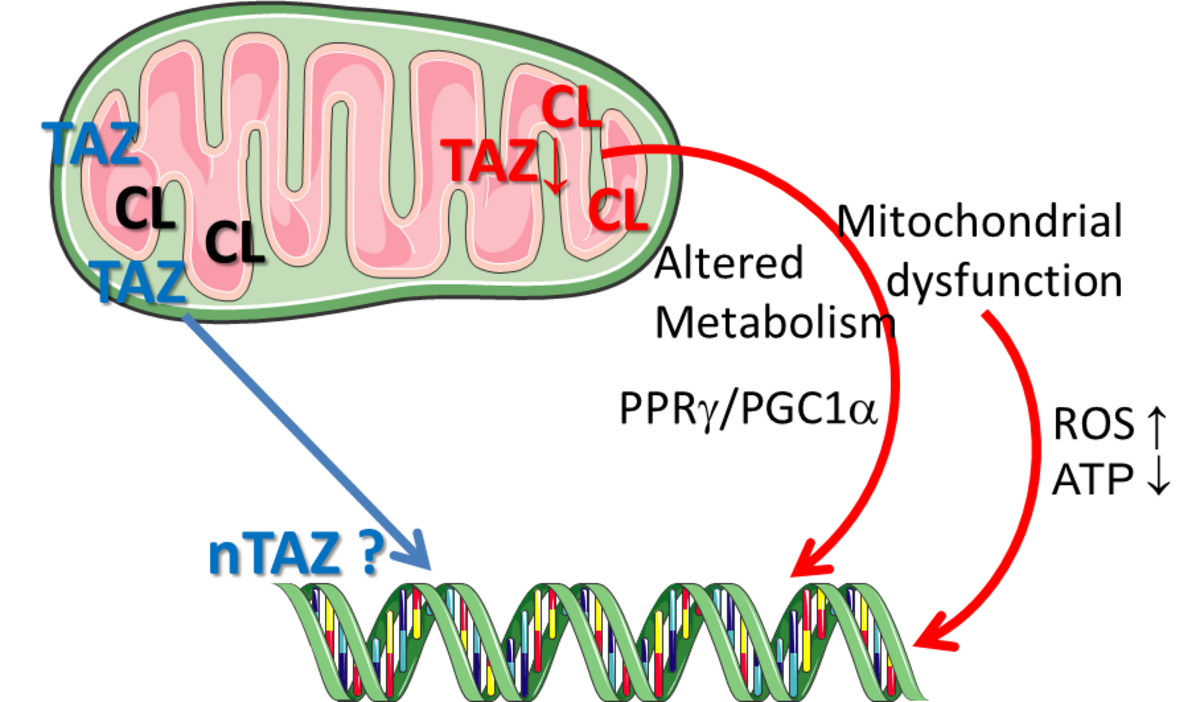
Tafazzin, a mitochondrial phospholipid-lysophospholipid transacylase, mediates the remodeling of cardiolipin (CL), a major phospholipid of the mitochondrial membranes[1]. Loss of function mutations in the human tafazzin (TAZ) cause the Barth syndrome[1], a disease that has been associated with compromised respiratory chain (RC) super-complex formation and, thus, increased electron leakage from the RC and reduced ATP production. Mitochondrial products like ATP and ROS (in particular O2- and H2O2) are key regulators of cellular metabolism and growth; however, physiologic mechanisms that control mitochondrial activity and translate it to cellular dysfunction in disease conditions and ageing remain incompletely understood[2]. In collaborative previous work we demonstrated that the knock-out of tafazzin ( TAZ-KO) not only alters mitochondrial membrane properties and decreases mitochondrial respiration, but also substantially compromises cell proliferation, cellular and mitochondrial morphology, cellular elasticity, ROS production/redox activity, and provokes rather specific changes in the expression of genes coding for receptors and proteases of the renin angiotensin system (RAS).
Proposed project
This project aims to identify molecular mechanisms that link Tafazzin-knock-out dependent alterations in mitochondrial structure (cardiolipin composition, membrane fluidity and elasticity) and function (respiration, OXPHOS) with cellular functions described above.
Main points that will be addressed are (I) to what extent the TAZ-KO mediated changes can be reversed by restoring the “wild-type” cardiolipin composition (by e.g. administration of selected fatty acids) and, (II) how the gene expression changes observed in both TAZ-KO or TAZ knock-down cells are mediated mechanistically.
A promoter analysis revealed that the affected genes share a consensus transcription factor binding site, which is also present in the promoter of the Fat1 gene. Fat1 has been suggested to link mitochondrial activity to cell growth. The pharmacological modulation of the signaling pathway activity targeting the shared consensus sites as well as the application of luciferase reporter-plasmids containing the native promoter sequence or systematically shortened promoter fragments, respectively, will facilitate the identification of mechanisms contributing to the TAZ-KO-mediated effects.
Mitochondrial structure and function will be determined in collaboration with Profs. Scholz and Helm and will be correlated to cardiolipin composition (Prof. Schild) and a broad variety of cellular functions (including ROS production/redox activity (PD Dr. Elsner) and cellular/mitochondrial elasticity (Dr. Otto). Tafazzin variants provided by Prof. Bornscheuer will be expressed in TAZ-KO cells. Correlating the resulting CL-composition with mitochondrial and cellular function will provide proof and mechanistic clues for a role of TAZ in cell growth.
This project will provide important insights into functions of tafazzin that are performed “down-stream” of cardiolipin remodeling.
Literature
[1] Schlame, M.; Greenberg, M. L. Biochim Biophys Acta 2017, 1862, 3-7.Cao LL, et al. Nature 2017, 539, 575-578.
Contact
Uwe Lendeckel
University Medicine Greifswald
Institute of Medical Biochemistry and Molecular Biology
Ferdinand-Sauerbruchstr.
D-17475 Greifswald
Tel: +49 (0)3834 86 5425
Fax:+49 (0)3834 86 5402
uwe.lendeckeluni-greifswaldde
Website