
Project A3
Doctoral researcher: Anika Wilden
Principle investigator: Matthias Sendler and Julia Mayerle
Co-supervisors: N.N.
Mitophagy-The thesis or antithesis of cell death in pancreatitis
Background
Acute pancreatitis, a fatal disease for 20% of severely affected patients, has been considered a disorder of pancreatic auto-digestion facilitated by premature and intracellular activation of digestive proteases in zymogens that induce tissue injury [1-5]. While zymogen activation has been considered the principal mechanism of injury, mitochondrial dysfunction has been associated increasingly consequential to intracellular calcium overload. Mitochondrial uptake of calcium drives normal cellular bioenergetics, but high calcium loads induce increasingly drastic responses culminating in necrosis [6]. Sustained intracellular calcium overload causes maintained elevations of reactive oxygen species (ROS), an inevitable by-product of mitochondrial metabolism, within the acinar cells [7].
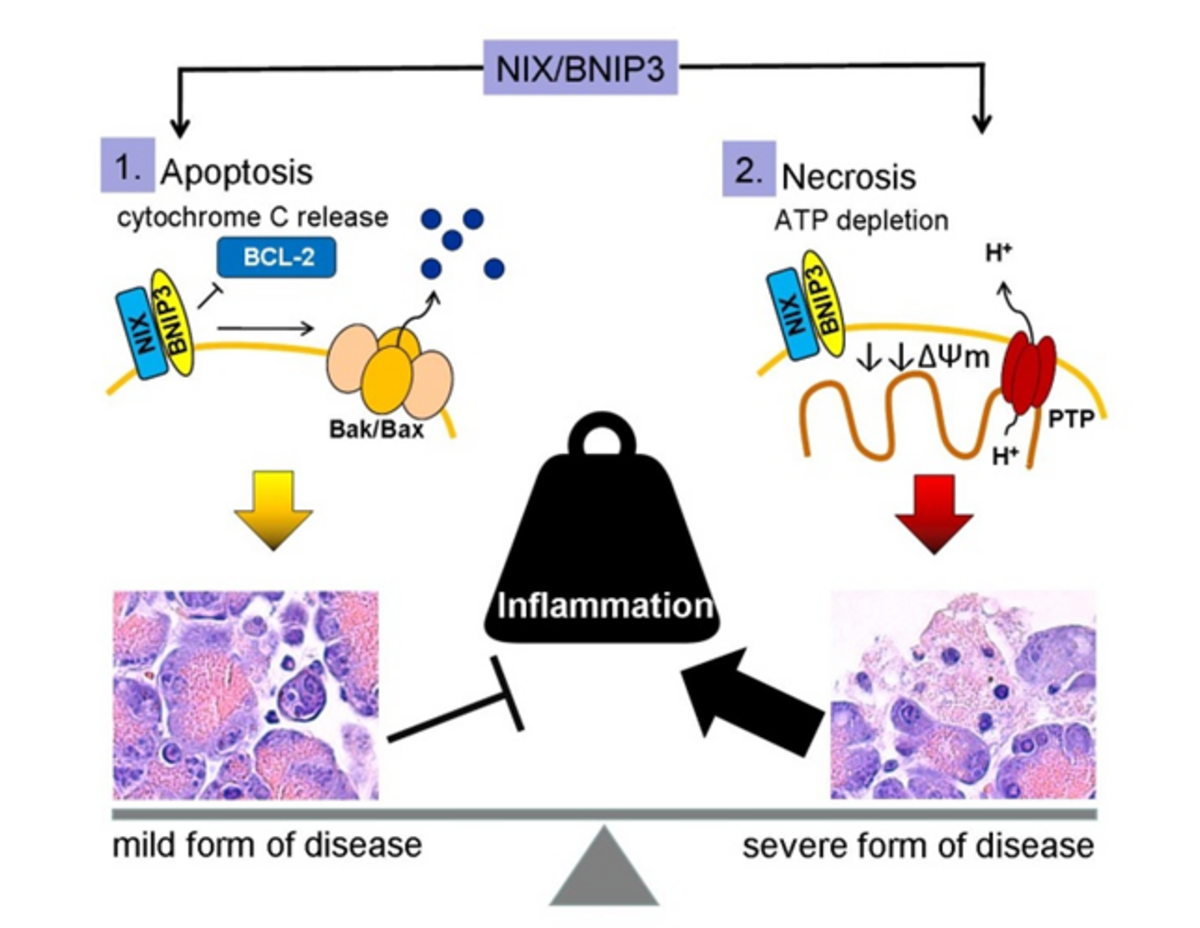
Also the mitochondrial membrane is destabilized during Ca2+ overload, which results in mitochondrial depolarization. The composition of mitochondrial membrane is essential for the maintenance of mitochondrial membrane potential and ATP generation. Oxidation of membrane components by ROS results in a destabilization of mitochondria and can induce apoptosis by cytochrome C release. To prevent cell death, autophagic degradation of damaged mitochondria is essential for cell survival. On the other hand, this decreases cellular ATP generation. A significant loss of ATP generation by massive mitochondrial depolarization leads to energy depletion and finally to necrotic cell death. The accumulation of damaged mitochondria is thought to be an inducer of cell death, therefore the selective removal of damaged mitochondria by autophagosomes and their subsequent catabolism by lysosomes, by a process called mitophagy, is essential for homeostasis of cells.
Mitophagy is known to be mediated by BNIP3 and BNIP3-like protein X[8]. This links mitochondrial membrane stability directly to cell death mechanisms, which defines the course of disease. Whereas apoptosis is an anti-inflammatory silent cell death, necrosis leads to the release of damage associated molecular patterns (DAMPs) associated with pro-inflammation.
BNIP3 (BCL2 and adenovirus E1B 19 kDa-interacting protein 3) and BNIP3-like (BNIP3L), also known as NIX, are proteins with homology to BCL2 in the BH3 domain, which induce both cell death and autophagy. BNIP3 and NIX localize to the outer mitochondrial membrane, where they function in mitophagy and mitochondrial dynamics by forming an isolating membrane around damaged mitochondria[9,10]. Consistent with their ability to induce cell death, BNIP3 and NIX are implicated in the pathogenesis of cancer and inflammatory disorders [11]. BNIP3 causes mitochondrial depolarization in cardiac myocytes, which is blocked by mitochondrial permeability transition pore (MPTP) inhibitors. BNIP3/NIX can cause cell death without cytochrome c release or caspase activation. The underlying mechanism is connected to activation of MPTP, increased ROS production and excessive autophagy[12]. Mitophagy can be detected in experimental pancreatitis [13] but the cellular consequence and consensual molecular events associated with BNIP3/NIX have not been studied. BNIP3/NIX could be a critical mechanism which determines cell death in pancreatitis by inducing apoptosis, necrosis or survival by removing damaged mitochondria by autophagy.
Hypothesis
- Stress induced overexpression of BNIP3/NIX in chronic pancreatitis leads to mitochondrial dysfunction and cell death.
- Alternatively, impairment of BNIP3/NIX functions leads to impaired mitochondrial quality control which in turn induces mitochondrial dysfunction and cell death.
- Deletion of BNIP3/NIX leads to elevated ROS production with subsequent inflammatory response and features of pancreatitis.
Methodology
To achieve our aim, we have set the following objectives:
- Elucidation of expression and compartmentalization of BNIP3, NIX, LC3, p62 and other autophagy markers in different models of pancreatitis (caerulein pancreatitis, L-arginine pancreatitis, bile duct ligation models) and comparison using GFP-LC3 transgenic mice, pancreatic acinar cell specific BNIP3-/-, and NIX-/- mice.
- To elucidate the role of mitochondrial dysfunction, mitophagy, ROS production, calcium influx in relation with BNIP3/NIX expression.
- To elucidate the role of programmed and un-programmed cell-death pathways in pancreatitis with respect to BNIP3/NIX signaling.
- Therapeutic intervention to ameliorate pancreatitis targeting BNIP3/NIX pathways using specific agonists (eg. HMOX induction, NAADP-AM, PMI), antagonists (eg. Fumonisin B1, NED-19) and mitochondria specific antioxidants (eg. N-acetyl-cysteine or ANTOX).
In all, these strategies will elucidate the overall molecular mechanism of BNIP3/NIX axis dependent mitophagy in pancreatitis. The choice of a model will allow to characterize the contribution of the different cell death mechanisms as well as the influence of mitochondrial dysfunction during the progression of pancreatitis.
Integration of the project into the existing structure of BiOx
As in the past we will strongly rely on the expertise of Professor Lendeckel in working on isolated mitochondria as well as on Dr. Elsner to use the HyPer protein for H2O2 sensing. The added benefit of this project within the GRK1947 will be the comparison of exocrine and endocrine pancreatic physiology with regard to ROS dependent mitophagy.
Literature
[1] Sendler, M. et al. Tumour necrosis factor α secretion induces protease activation and acinar cell necrosis in acute experimental pancreatitis in mice. Gut62, 430–439 (2013).
[2] Hohwieler M,et al. Humanpluripotent stem cell-derived acinar/ductal organoids generate human pancreasupon orthotopic transplantation and allow disease modelling. Gut.2016 Sep 15.pii: gutjnl-2016-312423. doi: 10.1136/gutjnl-2016-312423.
[3] Malla SR,et al Effect of oral administration of AZD8309, aCXCR2 antagonist, on the severity of experimental pancreatitis. Pancreatology.2016 Sep-Oct;16(5):761-9.
[4] Sendler M, Cathepsin B Activity InitiatesApoptosis via Digestive Protease Activation in Pancreatic Acinar Cells andExperimental Pancreatitis. J Biol Chem. 2016 Jul 8;291(28):14717-31.
[5] Leppkes M, et al. Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis. Nat Commun. 2016 Mar 11;7:10973.
[6] Mukherjee, R. et al.Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting theproduction of ATP. Gut65, 1333–1346 (2016).
[7] Criddle, D. N. Reactive oxygen species, Ca2+ stores and acute pancreatitis; a step closer to therapy? Cell Calcium60, 180–189 (2016).
[8] Youle, R. J. & Narendra, D. P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol.12, 9–14 (2011).
[9] Sandoval, H. et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature454, 232–235 (2008).
[10] Zhang, J. & Ney, P. A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ.16, 939–946 (2009).
[11] Ney, P. A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta1853, 2775–2783 (2015).
[12] Vande Velde, C. et al.BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol. Cell. Biol.20, 5454–5468 (2000).
[13] Jacob, T. G., Sreekumar, V. I., Roy, T. S. & Garg, P. K. Electron-microscopic evidence of mitochondriae containing macroautophagy in experimental acute pancreatitis: implications for cell death. Pancreatol. Off. J. Int. Assoc. Pancreatol. IAP Al14, 454–458 (2014).
Contact
Matthias Sendler
University Medicine Greifwald
Klinik für Innere Medizin A
Fleischmannstr. 41
D‐ 17475 Greifswald
Tel: +49 (0)3834 86 80096
Fax:+49 (0)3834 86 7234
sendlermuni-greifswaldde
Website
Julia Mayerle
Medizinische Klinik und Poliklinik II
Klinikum der LMU München-Grosshadern
Marchioninistr. 15
D‐ 81377 München
Tel: +49 (0)89 4400 72390
Fax:+49 (0)89 4400 78887
Julia.Mayerlemed.uni-muenchende
Website